- Classification of Thalassemia
- Pathophysiology of Thalassemia
- Clinical Manifestations of Thalassemia
- Diagnostic Approach to Thalassemia
- Modern Medical Treatment of Thalassemia
- Complications and Long-Term Outcomes
- Preventive Strategies and Genetic Counseling
- Ayurvedic Perspective and Integrative Care
- References
Thalassemia is a genetic blood disorder characterized by abnormal production of hemoglobin—the protein responsible for transporting oxygen within red blood cells. Derived from the Greek words thalassa (“sea”) and haima (“blood”), Thalassemia was initially recognized predominantly among populations surrounding the Mediterranean Sea, hence the historical term “Mediterranean anemia.” Today, Thalassemia is recognized as one of the most common inherited blood disorders worldwide, significantly impacting individuals of Middle Eastern, South Asian, Southeast Asian, African, and Mediterranean descent.
Globally, Thalassemia affects millions, with the highest prevalence in regions like India, Southeast Asia, and the Middle East. According to recent epidemiological estimates, approximately 80–90 million people worldwide are carriers of Thalassemia, while more than 300,000 individuals are born each year with severe forms of the disease (Weatherall, 2018).
From a physiological perspective, Thalassemia disrupts the body’s ability to produce sufficient healthy hemoglobin molecules. This defective hemoglobin production causes anemia, leading to symptoms ranging from mild fatigue and pallor in minor cases to severe anemia, organ enlargement, bone deformities, and growth failure in more severe forms. Without proper management, severe cases such as Beta Thalassemia Major—also known as Cooley’s anemia—can be life-threatening, highlighting the importance of early diagnosis and intervention.
To visualize the condition in simpler terms, imagine a factory designed to produce sturdy bricks for building homes. In Thalassemia, this factory, analogous to the bone marrow, consistently produces bricks (red blood cells) that are either incomplete or structurally weak. Consequently, these defective bricks fail to adequately support the structure—representing bodily functions dependent on oxygen supply—resulting in compromised overall health.
Early identification and regular medical intervention play crucial roles in mitigating Thalassemia’s impact on affected individuals, ensuring a better quality of life. Current treatments primarily focus on symptom management through regular blood transfusions, iron chelation therapies, and potentially curative approaches like bone marrow transplantation. Simultaneously, traditional medicine systems such as Ayurveda provide complementary approaches aimed at improving overall health and reducing disease severity through holistic, individualized treatment strategies.
In this comprehensive guide, we will explore the complexities of Thalassemia, integrating modern medical insights with Ayurvedic principles, to offer a holistic understanding that empowers both patients and healthcare providers.
Classification of Thalassemia
Thalassemia includes a spectrum of inherited blood disorders caused by mutations affecting hemoglobin chain production. It is classified based on whether the alpha or beta chains are deficient, and how many genes are involved.
Alpha Thalassemia arises from deletions in the HBA1 and HBA2 genes on chromosome 16. Since humans inherit two alpha-globin genes from each parent (total of four), the severity depends on how many genes are affected. A single gene deletion results in a silent carrier state—clinically asymptomatic but detectable through genetic testing. Deletion of two genes causes Alpha Thalassemia Trait, leading to mild microcytic anemia. Deletion of three genes leads to Hemoglobin H Disease, presenting with moderate anemia, jaundice, splenomegaly, and occasional bone changes. Complete deletion of all four genes results in Hydrops Fetalis, a fatal condition causing stillbirth due to severe anemia and generalized edema.
Beta Thalassemia is caused by mutations in the HBB gene on chromosome 11, resulting in reduced (β⁺) or absent (β⁰) synthesis of beta-globin chains. Individuals inherit one beta gene from each parent. When only one gene is mutated, the person has Beta Thalassemia Minor (Trait), which presents as mild microcytic anemia, often asymptomatic but with elevated HbA2 levels on electrophoresis. If both beta genes are mutated with mild variants, the result is Beta Thalassemia Intermedia, characterized by moderate anemia that may require occasional transfusions and carries a risk of iron overload from increased absorption. Homozygous severe mutations cause Beta Thalassemia Major (Cooley’s Anemia), leading to severe anemia manifesting in early infancy. These patients are transfusion-dependent and develop bone deformities, hepatosplenomegaly, and iron overload complications without curative intervention.
Other rare types include Delta-Beta Thalassemia, which combines deficiency of delta and beta chains and results in mild to moderate anemia, and E-beta Thalassemia, a compound heterozygous state combining beta-thalassemia and Hemoglobin E mutation, common in Southeast Asia with clinical severity ranging from intermediate to major depending on alleles.
For clarity, the following table summarizes the types of Thalassemia:

A real-life example involves a 5-year-old child from eastern India presenting with pallor, jaundice, and abdominal swelling. Lab tests showed hemoglobin 5.2 g/dL, MCV 62 fL, elevated HbF at 80%, and HbA2 at 6.2% on HPLC. Molecular analysis confirmed homozygous β⁰ mutation. The child was diagnosed with Beta Thalassemia Major and began regular transfusions and iron chelation therapy.
Reference:
Colah, R. B., Gorakshakar, A. C., & Nadkarni, A. H. (2010). Global burden, distribution and prevention of beta-thalassemias and hemoglobin E disorders. Expert Review of Hematology, 3(1), 103–117. https://doi.org/10.1586/ehm.09.74
Pathophysiology of Thalassemia

Thalassemia disrupts the body’s ability to produce functional hemoglobin, resulting in ineffective erythropoiesis and chronic anemia. The underlying pathology centers around an imbalance in globin chain synthesis, leading to a cascade of cellular and systemic effects.
In Beta Thalassemia, defective or absent synthesis of beta-globin chains causes an excess of unpaired alpha-globin chains. These free alpha chains are unstable and precipitate within developing erythroblasts in the bone marrow, forming intracellular inclusion bodies. The accumulated inclusions damage the erythroid precursors, leading to apoptosis and premature destruction—a process known as ineffective erythropoiesis. Consequently, despite increased erythropoietic activity, very few functional red blood cells enter the circulation.
In Alpha Thalassemia, depending on the number of gene deletions, there is a progressive deficiency of alpha-globin chains. The imbalance shifts toward excess beta or gamma chains. In cases like Hemoglobin H disease, excess beta chains form unstable tetramers (HbH) that have a high oxygen affinity but are ineffective in delivering oxygen to tissues. In the most severe form, Hydrops Fetalis, gamma chains aggregate into tetramers (Hemoglobin Bart’s), leading to severe hypoxia incompatible with fetal survival.
Both forms lead to chronic anemia, which stimulates increased erythropoietin production. This erythropoietic drive causes expansion of the bone marrow cavities (especially in the skull, maxilla, and long bones), manifesting as skeletal deformities and frontal bossing. The ineffective erythropoiesis also promotes iron absorption from the gastrointestinal tract, compounding iron overload, even in the absence of transfusions.
Another critical feature is extramedullary hematopoiesis, as the body attempts to compensate by producing blood cells outside the bone marrow, notably in the liver and spleen. This leads to hepatosplenomegaly, further complicating management.
On a molecular level, the abnormal red cells that escape the bone marrow exhibit reduced lifespan due to their membrane fragility and oxidative damage. They are prematurely cleared by the spleen, perpetuating the anemia.
A useful analogy is imagining a brick factory where one essential material (beta or alpha chains) is missing. As a result, incomplete or broken bricks accumulate inside the factory, causing machinery breakdown (ineffective erythropoiesis) and poor output (anemia), while the factory expands unsustainably to meet demand (marrow expansion), leading to structural deformities and further strain.
This pathophysiological cascade underscores why treatment in Thalassemia focuses not only on correcting anemia but also on controlling iron overload and preventing organ damage from chronic compensatory mechanisms.
Clinical Manifestations of Thalassemia

The clinical presentation of Thalassemia varies depending on the type and severity of the underlying genetic mutation. While individuals with minor forms may remain asymptomatic, those with more severe forms exhibit progressive, multisystem manifestations from early life.
Patients with Beta Thalassemia Major typically present between 6 to 12 months of age, once fetal hemoglobin declines and defective adult hemoglobin production becomes evident. Common symptoms include profound fatigue, pallor, and failure to thrive. The hallmark of the disease is severe anemia requiring regular transfusions.
As ineffective erythropoiesis continues, the bone marrow expands in an attempt to compensate, leading to characteristic skeletal changes such as frontal bossing, prominent cheekbones, and maxillary overgrowth causing dental malocclusion. Radiographs may reveal a “hair-on-end” appearance of the skull due to marrow hyperplasia.
Hepatosplenomegaly develops as extramedullary hematopoiesis shifts to the liver and spleen. Splenomegaly can worsen anemia by increasing red cell sequestration, leading to a vicious cycle of anemia and enlargement. Progressive enlargement may cause early satiety and abdominal discomfort.
Iron overload arises both from increased intestinal absorption (due to ineffective erythropoiesis) and recurrent transfusions. Over time, iron accumulates in critical organs including the heart, liver, and endocrine glands. This can result in complications such as cardiac hemosiderosis (leading to heart failure), hepatic fibrosis and cirrhosis, diabetes mellitus (due to pancreatic iron deposition), and hypogonadotropic hypogonadism causing delayed or absent puberty.
Children with Alpha Thalassemia Trait or Beta Thalassemia Minor are usually asymptomatic but may show mild microcytic anemia on routine blood testing. In Hemoglobin H disease, patients may experience mild to moderate anemia, jaundice, and splenomegaly, occasionally requiring transfusions during stress or infections.
A real-life example involves a 7-year-old boy from the Mediterranean region presenting with progressive pallor, irritability, abdominal distension, and frontal bossing. Examination revealed severe anemia (Hb 5.4 g/dL), palpable hepatosplenomegaly, and characteristic facial bone changes. Hemoglobin electrophoresis confirmed Beta Thalassemia Major with elevated HbF and absence of HbA. He was initiated on a regular transfusion protocol and iron chelation therapy to prevent long-term complications.
The psychological impact of Thalassemia must also be acknowledged. Children and adolescents undergoing lifelong transfusions and monitoring may develop emotional distress, social withdrawal, or academic difficulties due to frequent hospitalizations and visible physical differences.
The constellation of anemia, skeletal deformities, organomegaly, and iron-related complications underscores the need for a multidisciplinary approach in managing Thalassemia, aiming not only at correcting anemia but also at preventing the progression of end-organ damage and preserving quality of life.
Diagnostic Approach to Thalassemia

The diagnosis of Thalassemia involves a combination of clinical evaluation, hematological testing, and genetic analysis to confirm the type and severity of the disorder.
Initial suspicion arises in children presenting with chronic anemia, pallor, jaundice, or hepatosplenomegaly, particularly in regions with high Thalassemia prevalence or a family history of the disease.
The diagnostic workup begins with a complete blood count (CBC). This typically shows microcytic hypochromic anemia (low mean corpuscular volume [MCV], low mean corpuscular hemoglobin [MCH]), with a normal or elevated red blood cell count—helping differentiate Thalassemia from iron deficiency anemia. Peripheral blood smear reveals target cells, anisopoikilocytosis, and nucleated red blood cells.
Reticulocyte count is normal or mildly elevated, reflecting ineffective erythropoiesis. Serum iron and ferritin levels are usually normal or increased, further distinguishing it from iron deficiency.
A cornerstone of diagnosis is hemoglobin electrophoresis or high-performance liquid chromatography (HPLC) to quantify hemoglobin fractions. In Beta Thalassemia Minor, HbA2 is elevated (>3.5%), while in Beta Thalassemia Major, HbA is absent or markedly reduced with elevated HbF (>90%). Alpha Thalassemia Trait typically shows normal electrophoresis; diagnosis requires molecular analysis.
When Alpha Thalassemia is suspected, DNA-based tests such as gap-PCR or multiplex ligation-dependent probe amplification (MLPA) identify alpha-globin gene deletions. For Beta Thalassemia, sequencing the HBB gene confirms point mutations or small insertions/deletions.
For prenatal diagnosis, chorionic villus sampling or amniocentesis combined with molecular testing allows early detection in at-risk pregnancies. Carrier screening using HPLC or electrophoresis in parents is critical for genetic counseling.
Additional investigations include:
- Serum ferritin and liver iron quantification (MRI T2)* to monitor iron overload in transfused patients.
- Bone marrow examination (rarely needed) showing erythroid hyperplasia.
- Radiographs may show characteristic skeletal changes (hair-on-end skull appearance).
Differential diagnoses include iron deficiency anemia, sideroblastic anemia, lead poisoning, and anemia of chronic disease. The combination of microcytic anemia, normal iron stores, elevated RBC count, and abnormal hemoglobin fractions strongly supports Thalassemia.
Early and accurate diagnosis not only enables optimal treatment planning but also facilitates family counseling and prevention through carrier detection and prenatal screening.
A real-life case involved a 9-month-old infant with progressive pallor, jaundice, and abdominal swelling. Blood tests showed Hb 6.1 g/dL, MCV 65 fL, and target cells on smear. HPLC revealed HbF 85%, HbA absent, HbA2 4%. Genetic testing confirmed homozygous β⁰ mutation, establishing a diagnosis of Beta Thalassemia Major.
Modern Medical Treatment of Thalassemia

The treatment of Thalassemia is centered on correcting chronic anemia, preventing iron overload, and minimizing disease-related complications. Although curative therapies remain limited, advances in supportive care have dramatically improved both survival and quality of life for individuals living with Beta Thalassemia Major and other severe forms.
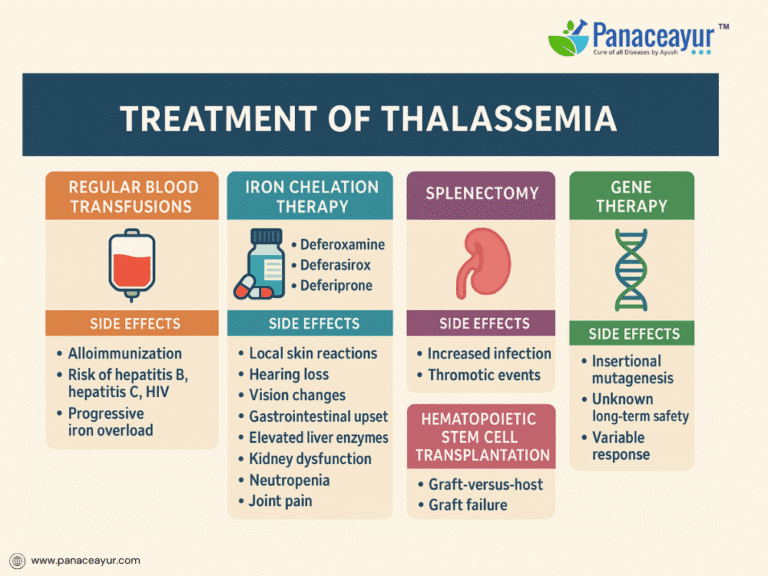
Regular blood transfusions remain the cornerstone of treatment, particularly for Beta Thalassemia Major. Lifelong transfusions, initiated in early childhood, aim to maintain pre-transfusion hemoglobin levels between 9 and 10 g/dL. This level effectively suppresses ineffective erythropoiesis, supports normal growth, and prevents skeletal deformities. Despite their lifesaving role, transfusions carry inherent risks. Patients may develop alloimmunization, forming antibodies against donor red cells that complicate future transfusions. Transfusion-transmitted infections, though minimized through rigorous screening, remain a concern for hepatitis B, hepatitis C, and HIV. Each transfusion introduces an iron load of approximately 200–250 mg, progressively contributing to iron accumulation. Early and extended red cell antigen typing is recommended to reduce the risk of alloimmunization, especially in transfusion-dependent patients.
Iron overload, a direct consequence of transfusion dependency, is inevitable without intervention. Excess iron deposits in vital organs such as the liver, heart, and endocrine glands, leading to progressive complications including cardiac failure, liver fibrosis, and endocrine dysfunction. Chelation therapy is essential to remove excess iron and protect against organ damage. Deferoxamine, administered as a subcutaneous or intravenous infusion over 8 to 12 hours for 5 to 7 nights per week, has been a mainstay of chelation. However, it is associated with notable side effects, including local skin reactions, hearing loss, vision impairment, and skeletal abnormalities such as bone dysplasia and growth retardation. The need for prolonged nightly infusions via pump significantly affects patient adherence and quality of life.
Oral chelators offer improved convenience and compliance. Deferasirox is taken once daily on an empty stomach and carries a side effect profile including gastrointestinal disturbances, elevated liver enzymes indicating hepatotoxicity, kidney dysfunction manifesting as proteinuria or rising creatinine, and occasional skin rashes. Regular liver and kidney function monitoring is critical during therapy. Deferiprone, another oral chelator requiring thrice-daily dosing, introduces the risk of neutropenia and potentially life-threatening agranulocytosis, necessitating weekly white blood cell counts. Joint pain, gastrointestinal symptoms, and elevated liver enzymes are additional adverse effects. In some cases, deferiprone is combined with deferoxamine for synergistic iron removal, particularly in patients with cardiac iron overload.
Splenectomy may be considered for patients with massive splenomegaly causing discomfort, hypersplenism resulting in excessive red cell destruction, or rising transfusion needs. However, splenectomy carries its own risks, including heightened susceptibility to life-threatening infections by encapsulated bacteria such as pneumococcus, meningococcus, and Haemophilus influenzae. It also increases the risk of thrombotic events such as portal vein thrombosis and pulmonary embolism. Therefore, splenectomy candidates require preoperative vaccination and, in some cases, lifelong antibiotic prophylaxis to mitigate infection risks.
For select patients, hematopoietic stem cell transplantation (HSCT) offers the potential for a lifelong cure. Ideally performed in childhood using an HLA-matched sibling donor, HSCT can free patients from transfusions and chelation. However, this procedure is not without significant risks, including graft-versus-host disease, graft failure, severe infections related to immunosuppression, and organ toxicity from conditioning regimens. Outcomes are best in younger patients without preexisting iron overload or liver damage, making early referral and preparation critical.
Exciting advancements in gene therapy are offering new hope. Clinical trials using lentiviral vectors to introduce functional beta-globin genes into a patient’s own hematopoietic stem cells have shown promising results, with some individuals achieving transfusion independence. Nonetheless, gene therapy remains experimental, with concerns about insertional mutagenesis and unknown long-term safety. Parallel research in gene editing technologies, such as CRISPR-Cas9, is targeting regulators of fetal hemoglobin to compensate for deficient adult hemoglobin production, though these approaches are still in investigational stages.
In addition to disease-specific treatments, supportive care plays a critical role. Comprehensive monitoring includes regular assessment of growth, pubertal development, endocrine function, and bone health to detect and manage complications such as diabetes, hypothyroidism, hypogonadism, and osteoporosis. Patients benefit from a multidisciplinary care approach involving hematology, endocrinology, cardiology, and psychosocial support services to address the multifaceted impact of chronic disease.
A real-life case involves a 15-year-old adolescent receiving deferasirox chelation therapy who developed rising liver enzymes and proteinuria, necessitating a dose adjustment and close monitoring of hepatic and renal function. Transitioning this patient to adult care required coordinated management across specialties, including cardiology and endocrinology, to monitor cardiac iron load, hormone status, and long-term organ health.
Modern treatment of Thalassemia has transformed a once fatal pediatric disease into a manageable chronic condition. Nevertheless, lifelong adherence to complex treatment regimens and vigilance for therapy-related side effects remain essential to preserve quality of life and prevent long-term complications.
Complications and Long-Term Outcomes

Despite advancements in treatment, Thalassemia remains associated with significant long-term complications due to chronic anemia, iron overload, and treatment side effects. These complications impact multiple organ systems and underscore the need for lifelong monitoring and multidisciplinary care.
Iron overload, whether from transfusions or increased intestinal absorption, is the primary driver of morbidity in transfusion-dependent patients. Excess iron deposits progressively in the liver, leading to hepatomegaly, hepatic fibrosis, and eventually cirrhosis. In the heart, iron deposition causes cardiomyopathy, manifesting as diastolic dysfunction progressing to congestive heart failure, a leading cause of mortality in Thalassemia Major. Endocrine complications result from iron-induced damage to the pituitary, pancreas, and thyroid, leading to hypogonadotropic hypogonadism, delayed puberty, infertility, hypothyroidism, and diabetes mellitus.
Skeletal complications arise from marrow expansion, particularly in undertreated or poorly transfused patients. Chronic ineffective erythropoiesis leads to bone marrow hyperplasia, causing characteristic skeletal deformities such as frontal bossing, maxillary overgrowth, and thinning of cortical bone, predisposing to fractures and osteoporosis. Radiological findings often include “hair-on-end” appearance of skull bones and widened medullary spaces in long bones.
Splenomegaly is a common consequence of extramedullary hematopoiesis and may lead to early satiety, discomfort, and increased red cell sequestration, further exacerbating anemia. Hypersplenism can worsen thrombocytopenia and leukopenia, increasing the risk of infections and bleeding. Post-splenectomy patients face lifelong increased susceptibility to severe infections from encapsulated bacteria, necessitating vaccination and prophylactic measures.
Endocrine complications extend to growth failure and bone mineral density loss. Many adolescents experience delayed or absent secondary sexual development due to hypogonadism. Osteoporosis and osteopenia are prevalent, partly due to iron toxicity in bone and hormonal deficiencies, increasing fracture risk.
Gallstones, often pigmented due to chronic hemolysis, are another complication, leading to cholecystitis or biliary colic. Chronic anemia also increases the risk of leg ulcers and thrombotic events, including pulmonary embolism and portal vein thrombosis, particularly in patients post-splenectomy.
Psychosocial complications are frequently overlooked but critically impact quality of life. Patients may experience anxiety, depression, poor self-esteem related to physical appearance (skeletal changes, short stature), and social stigma. Academic and employment challenges arise due to frequent hospitalizations and treatment burden.
The cumulative burden of these complications can reduce life expectancy and quality of life if not adequately managed. However, with optimal transfusion regimens, effective chelation, early detection of complications, and multidisciplinary care, survival into adulthood with preserved organ function is increasingly achievable. Regular screening using liver and cardiac MRI, bone density assessments, endocrine evaluations, and psychosocial support form the foundation of modern Thalassemia care.
A 22-year-old woman with Beta Thalassemia Major, well-chelated since childhood, presented with secondary amenorrhea and osteoporosis. Endocrine evaluation revealed hypogonadotropic hypogonadism requiring hormone replacement therapy. Her cardiac MRI showed mild myocardial iron overload, prompting chelation intensification. Despite these complications, she maintained normal liver function and preserved fertility options through assisted reproductive counseling.
Long-term outcomes are closely tied to the degree of iron overload control and early identification of organ dysfunction. Evolving therapies, including gene therapy, hold the promise of modifying the natural history of Thalassemia, potentially reducing long-term morbidity in future generation
Preventive Strategies and Genetic Counseling
Preventing the birth of children affected by severe forms of Thalassemia has been a major public health focus in regions with high disease prevalence. Advances in carrier screening, prenatal diagnosis, and reproductive options have made it possible to significantly reduce new cases when integrated into national health programs and individual counseling.
Thalassemia follows an autosomal recessive inheritance pattern. Couples who are both carriers of a β-thalassemia mutation have a 25% chance with each pregnancy of having an affected child, a 50% chance of having a carrier child, and a 25% chance of having an unaffected, non-carrier child. Awareness of carrier status is therefore critical in reproductive decision-making.
Carrier screening is typically performed using complete blood count and hemoglobin electrophoresis or HPLC in individuals from at-risk populations or during premarital or preconception counseling. A low mean corpuscular volume (MCV) and elevated HbA2 (>3.5%) are suggestive of Beta Thalassemia Trait. Molecular genetic testing confirms the specific mutation, especially in equivocal cases or Alpha Thalassemia where electrophoresis may be normal.
Genetic counseling provides prospective parents with an understanding of inheritance risks, reproductive options, and implications of Thalassemia. It supports informed decision-making while respecting cultural, ethical, and personal values. Counseling sessions explain disease severity, treatment burden, quality of life impacts, and available preventive interventions.
Prenatal diagnosis is a cornerstone of prevention. Techniques such as chorionic villus sampling (CVS) at 10–12 weeks or amniocentesis at 15–18 weeks enable fetal genotyping when parental mutations are known. DNA analysis determines whether the fetus has inherited no mutation, is a carrier, or is affected. Couples can then consider pregnancy continuation or termination based on their values and the diagnosis.
In vitro fertilization (IVF) with preimplantation genetic diagnosis (PGD) offers an alternative to prenatal testing. Embryos are tested for the known parental mutations before implantation, allowing selection of unaffected or carrier embryos for transfer. PGD avoids the ethical and emotional complexities of termination but requires advanced laboratory facilities, higher costs, and may not be universally accessible.
In communities with high carrier prevalence, premarital screening programs combined with public education have been effective in reducing new Thalassemia births, notably in Cyprus, Iran, and Sardinia. Such programs must be paired with counseling to ensure understanding and avoid stigmatization.
Family cascade screening also plays a critical role. Identifying carriers within the extended family facilitates early counseling and testing in siblings, cousins, and other relatives, supporting broader prevention efforts.
While preventive strategies have proven highly effective, they raise ethical, cultural, and religious considerations requiring sensitive handling by genetic counselors and healthcare providers. Balancing individual autonomy with community health goals remains a key challenge.
A case study from Cyprus demonstrated the impact of national prevention programs. Following implementation of universal premarital screening and counseling in the 1970s, the number of births of children with Thalassemia Major fell from over 50 per year to nearly zero within two decades, without coercive measures, highlighting the power of informed reproductive choice.
Preventive strategies are integral to comprehensive Thalassemia management, reducing disease burden for families and health systems while enabling at-risk couples to make empowered reproductive decisions.
Ayurvedic Perspective and Integrative Care

Thalassemia, a genetically inherited disorder affecting hemoglobin synthesis, presents a unique interpretive challenge within Ayurveda. While classical texts do not define Thalassemia as a singular disease, its clinical features closely align with the conceptual framework of Raktavaha Srotas Dushti (disorders of the blood-carrying channels), Rakta Dhatu Kshaya (deficiency of blood tissue), and Beejadosha (congenital or hereditary defects transmitted through defective reproductive material). Ayurveda recognizes the influence of Beeja (genetic seed), Beeja Bhaga (genetic component), and Beeja Bhaga Avayava (subcomponents of genetic material) in determining the blueprint of health at conception. Under this paradigm, Thalassemia may be categorized as a Beejadosha Janya Vyadhi, reflecting a fundamental defect originating at the level of the genetic seed, manifesting primarily in the Rakta Dhatu. This philosophical understanding parallels modern science’s attribution of Thalassemia to mutations in the HBA1, HBA2, or HBB genes leading to alpha or beta chain deficiencies.
In Ayurveda, Rakta Dhatu is synthesized through the progressive transformation of Ahara Rasa (nutritive fluid derived from digested food) by Dhatvagni (tissue-specific metabolic fire). Any deficiency, distortion, or inefficiency in this chain—whether due to Agni Mandya (digestive/metabolic weakness), Rasa Dushti (impurity in primary nutrient), or Dhatvagni Dushti (tissue metabolic dysfunction)—results in Rakta Kshaya, manifesting clinically as pallor, fatigue, weakness, and dyspnea, collectively described under Pandu Roga. In the context of Thalassemia, however, the deficiency transcends mere quantity; it reflects a structural and functional defect in the very quality of Rakta Dhatu. The chronic destruction of immature red cells (ineffective erythropoiesis) mirrors Dhatu Paka, or degenerative pathology of tissues, further weakening systemic vitality.
The compensatory expansion of bone marrow observed in Thalassemia parallels the Ayurvedic understanding of Uttara Uttara Dhatu Anukarana, where upstream tissues attempt to fulfill the deficient function of downstream tissues. The resultant skeletal deformities, hepatosplenomegaly, and medullary expansion are manifestations of compounded Rakta, Meda, and Asthi Dhatu Vikriti, showing that the pathological process involves deeper tissue levels beyond the blood itself. The splenomegaly reflects heightened activity of Rakta-Meda Srotas as the body redirects hematopoietic processes into extramedullary sites.
Ayurvedic management does not claim to cure Thalassemia’s genetic root, yet offers supportive strategies to enhance tissue resilience, delay complications, improve metabolic efficiency, and improve quality of life. Its therapeutic goals include strengthening Rakta Dhatu formation, improving Agni, protecting the liver and spleen, reducing iron-induced oxidative stress, and minimizing side effects of conventional treatments. This is achieved through the judicious use of Rasayana Dravyas (rejuvenative agents), iron-assimilative formulations, and organ-protective interventions. Mandura Bhasma, a classical iron calx processed with Triphala decoction, is cited in Bhaishajya Ratnavali as a Rakta Janaka and Yakrit Pliha Rasayana. Punarnava Mandura, combining Mandura Bhasma with Punarnava, Trikatu, and Triphala, enhances liver function, reduces inflammation, and improves hematopoiesis. Navayasa Lauh and Lohasava are similarly employed to enhance iron assimilation, digestion, and mild hepatoprotection. Rasayana herbs like Guduchi (Tinospora cordifolia), Triphala, Amalaki (Emblica officinalis), and Ashwagandha (Withania somnifera) further contribute antioxidant, immunomodulatory, and adaptogenic benefits, which modern research corroborates as hepatoprotective and anti-inflammatory.
A 2015 review by Kapoor et al. demonstrated the role of herbal compounds such as Guduchi and Triphala in mitigating iron-induced hepatotoxicity in experimental models, reinforcing their relevance in managing oxidative stress in Thalassemia. Panchakarma therapies, notably Basti (medicated enema), may be selectively employed to balance Vata, strengthen Agni, and support tissue nourishment, while Nasya (nasal administration) may aid neuroendocrine regulation. However, aggressive detoxification is contraindicated in fragile patients; personalized protocols based on Prakriti (constitution), Bala (strength), and Vyadhi Avastha (disease stage) are essential.
Dietary recommendations focus on Rakta-enhancing foods such as pomegranate, black sesame, jaggery, beetroot, dates, and leafy greens, always tailored to digestive strength to prevent Mandagni or Ama formation. Patients are advised to avoid excessive sour, salty, and fermented foods that aggravate Pitta and burden liver metabolism. Lifestyle guidance integrates yoga, pranayama (particularly Anulom Vilom for balancing Vata-Pitta), and stress reduction, acknowledging the psychosomatic dimensions of chronic illness. Maintaining Satva Bala, or mental resilience, is emphasized as essential for sustaining long-term therapeutic engagement.
Integrative care positions Ayurvedic interventions alongside modern transfusion and chelation protocols to support liver function, minimize fatigue, enhance appetite, improve immunity, and mitigate the psychosocial toll of chronic therapy. A case reported by Agarwal & Goel (2010) described a Thalassemia Minor patient managed with Punarnava Mandura and Guduchi showing improved hemoglobin and reduced fatigue over six months, exemplifying Ayurveda’s supportive role. Philosophically, Ayurveda’s approach focuses less on correcting a defective gene and more on harmonizing the internal milieu, creating an environment where the body functions with maximum efficiency despite inherent genetic limitations.
This integrative model reflects a complementary paradigm, where modern medicine provides molecular-level interventions while Ayurveda enhances the patient’s systemic balance, resilience, and capacity to cope with the demands of lifelong therapy.
References
- Christou, S., & Kleanthous, M. (2020). Prevention of beta-thalassemia in Cyprus: A 35-year success story. Haematologica, 105(1), 19–22. https://doi.org/10.3324/haematol.2019.233387
- Kapoor, S., Chopra, H., & Khan, A. (2015). Herbal remedies in the management of iron overload: A review. Journal of Pharmacognosy and Phytochemistry, 4(1), 186–190. https://www.phytojournal.com/archives/2015/vol4issue1/PartC/4-1-15.pdf
- Taher, A. T., Musallam, K. M., Cappellini, M. D., & Weatherall, D. J. (2021). Optimal management of beta thalassemia: Current perspectives. Orphanet Journal of Rare Diseases, 16(1), 1–13. https://doi.org/10.1186/s13023-021-01973-0
- Taher, A. T., Saliba, A. N., & Cappellini, M. D. (2021). Thalassemias. The Lancet, 397(10291), 369–383. https://doi.org/10.1016/S0140-6736(20)01363-7
- Agarwal, R., & Goel, S. (2010). Ayurvedic perspective of anemia and its management. AYU, 31(4), 472–478. https://doi.org/10.4103/0974-8520.82039
- Sharma, P. V. (2006). Charaka Samhita: Chikitsa Sthana (16th Chapter). Chaukhambha Orientalia.
- Pandey, G. S. (2004). Bhaishajya Ratnavali: Pandu Chikitsa Adhyaya. Chaukhambha Sanskrit Sansthan.
- Bhavaprakasha Nighantu. (1999). Haritakyadi Varga. Chaukhambha Bharti Academy.
- Weatherall, D. J. (2010). The inherited diseases of hemoglobin are an emerging global health burden. Blood, 115(22), 4331–4336. https://doi.org/10.1182/blood-2010-01-251348
- De Sanctis, V., Soliman, A., Yassin, M., & Elsedfy, H. (2017). Bone disease in thalassemia: A frequent and still unresolved problem. Journal of Bone Metabolism, 24(4), 195–200. https://doi.org/10.11005/jbm.2017.24.4.195
- Borgna-Pignatti, C., & Galanello, R. (2004). Thalassemias and related disorders: Quantitative disorders of hemoglobin synthesis. In R. A. Greer et al. (Eds.), Wintrobe’s Clinical Hematology (11th ed., pp. 1319–1365). Lippincott Williams & Wilkins.
- Cappellini, M. D., Cohen, A., Porter, J., Taher, A., & Viprakasit, V. (2014). Guidelines for the management of transfusion dependent thalassaemia (TDT) (3rd ed.). Thalassaemia International Federation. https://thalassaemia.org.cy/publications/
- Porter, J. B. (2001). Practical management of iron overload. British Journal of Haematology, 115(2), 239–252. https://doi.org/10.1046/j.1365-2141.2001.03077.x
- Elalfy, M. S., Adly, A. A. M., Ismail, E. A., & Tantawy, A. A. G. (2014). Soluble transferrin receptor: A reliable marker for iron overload in children with beta-thalassemia major. Hematology, 19(4), 203–207. https://doi.org/10.1179/1607845413Y.0000000097
- Vichinsky, E. P. (2005). Clinical applications of chelation therapy. American Journal of Hematology, 80(6), 431–439. https://doi.org/10.1002/ajh.20403
- Ayyappan, R., Krishnan, R., & Rajalakshmi, K. (2008). Hepatoprotective and antioxidant activities of Triphala Churna in iron overload-induced hepatotoxicity in albino rats. Indian Journal of Pharmacology, 40(5), 236–240. https://www.ijph.in/article.asp?issn=0253-7613;year=2008;volume=40;issue=5;spage=236;epage=240;aulast=Ayyappan
- Karagiorga-Lagana, M. (2001). Genetic counselling in hemoglobinopathies: The Greek experience. Community Genetics, 4(1), 27–35. https://doi.org/10.1159/000046854
- Lad, V. (1994). Textbook of Ayurveda: Fundamental Principles. The Ayurvedic Press.
- Rao, R. V., & Bapat, R. D. (1987). Role of Ayurvedic drugs in liver diseases. Ancient Science of Life, 6(4), 236–241. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3336654/
- Maheshwari, R. K., & Singh, S. (2012). Rasayana: Ayurvedic herbs for longevity and rejuvenation. Ancient Science of Life, 31(3), 139–142. https://doi.org/10.4103/0257-7941.107361